Biosimilars and generics are both affordable alternatives to brand-name medications. These drugs play a crucial role in expanding patient access to life-saving treatments. While both generics and biosimilars serve the same purpose, they represent fundamentally different products with distinct regulatory pathways. Understanding these differences is essential for pharmaceutical sponsors navigating the complex approval process and identifying strategic opportunities in drug development.
Let’s dive into how biosimilars and generics are different from each other.
What Sets Biosimilars Apart from Generics
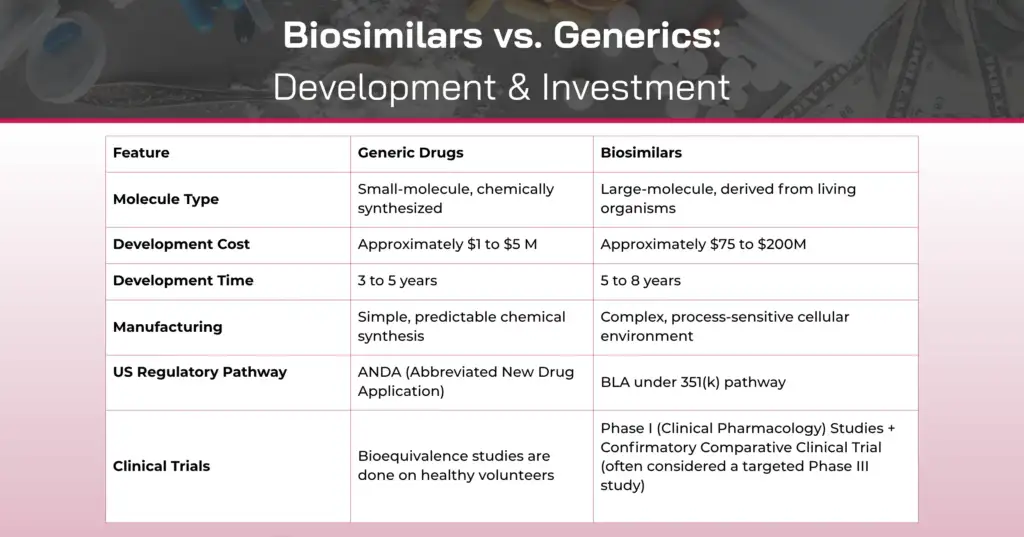
At their core, the distinction between biosimilars and generics stems from molecular complexity and manufacturing processes.
Generic drugs are typically small-molecule compounds that are chemically synthesized. The manufacturing process produces an active ingredient that is identical within each manufactured lot and between lots.
Biosimilars, in contrast, are biological products manufactured from living systems such as microorganisms, yeast, bacteria, or animal cells. These are large, complex molecules like monoclonal antibodies, therapeutic proteins, or vaccines. Because they originate from living systems, inherent molecular variation naturally occurs within each lot and between lots during the manufacturing process. This variation includes minor changes to the protein molecule, such as differences in glycosylation patterns, which are a natural part of biological manufacturing.
This fundamental difference in origin and complexity shapes everything from manufacturing controls to regulatory requirements.
Regulatory Standards: Bioequivalence vs. Biosimilarity
The approval standards for generics and biosimilars reflect their distinct molecular characteristics.
Generic Drug Approval
For generic drug approval under an Abbreviated New Drug Application (ANDA), manufacturers must demonstrate:
- Bioequivalence to the reference listed drug (RLD)
- That the generic contains the same active ingredient as the brand-name drug
- That it has the same strength, dosage form, and route of administration
- That it meets the same quality
The bioequivalence requirement means the generic drug performs in the same manner as the brand-name drug, releasing the active ingredient into the bloodstream at the same rate and extent. This is typically demonstrated through comparative pharmacokinetic studies in healthy volunteers.
Biosimilar Approval
Biosimilar approval under Section 351(k) of the Public Health Service Act requires manufacturers to demonstrate:
- High similarity to the reference biological product, notwithstanding minor differences in clinically inactive components
- No clinically meaningful differences between the biosimilar and reference product in terms of safety, purity, and potency
This is a more comprehensive evaluation than a generic bioequivalence. It includes extensive analytical characterization, comparative clinical studies demonstrating similar efficacy and safety profiles, and rigorous assessment of immunogenicity potential.
The Filing Process for Generics: The ANDA Pathway
The Abbreviated New Drug Application pathway for generics is designed to streamline approval by relying on the safety and efficacy data of the reference listed drug.
Key Steps in Generic Drug Development and Filing:
1. Identification of the Reference Product: Select the brand-name drug to be replicated and confirm its regulatory status
2. Formulation Development: Develop a formulation that matches the reference drug in strength, dosage form, and route of administration
3. Analytical Testing: Conduct dissolution testing and analytical studies to demonstrate pharmaceutical equivalence
4. Bioequivalence Studies: Perform comparative pharmacokinetic studies, typically in healthy volunteers, to demonstrate that the generic delivers the same amount of active ingredient to the bloodstream in the same timeframe as the reference drug
5. ANDA Preparation and Submission: Compile the application, including:
- Chemistry, manufacturing, and controls (CMC) information
- Bioequivalence study data
- Labeling that mirrors the reference product
- Patent certifications (Paragraph I, II, III, or IV)
6. Regulatory Submission and Approval
Finally, regulatory dossiers like ANDA (Abbreviated New Drug Application) or DCP/MRP are submitted to authorities such as the US FDA or EMA. These dossiers contain all clinical, technical, and manufacturing data to seek market authorization.
Patent Challenges: The Para IV Opportunity
A critical strategic consideration for generic manufacturers is the patent certification. Under a Paragraph IV certification, a generic applicant declares that the patents listed in the FDA’s Orange Book are invalid, unenforceable, or will not be infringed by the manufacture, use, or sale of the generic drug.
This certification triggers a 30-month stay during which the brand manufacturer can challenge the generic in court. However, the first generic applicant to file an ANDA with a Paragraph IV certification and successfully challenge the patent may be eligible for 180 days of market exclusivity. It provides a significant commercial advantage, allowing the first generic to market before competitors can enter.
The Filing Process for Biosimilars: The 351(k) BLA Pathway
A 351(k) application is what a company submits to the FDA to get approval for a biosimilar — a biological product that is highly similar to an already approved reference biologic.
To be approved, the 351(k) application must show that the biosimilar:
- It is highly similar to the reference product, with only minor differences in inactive ingredients.
- Uses the same mechanism of action (if known) as the reference product.
- It is approved for the same conditions of use listed in the reference product’s label.
- Has the same route of administration, dosage form, and strength.
- It is made in a facility that meets FDA standards for safety, purity, and potency.
The application must include data from:
- Analytical studies proving high similarity.
- Animal studies assessing toxicity (if needed).
- Clinical studies evaluating safety, immunogenicity, and how the drug behaves in the body (PK/PD studies).
The FDA may decide that some studies are unnecessary if sufficient evidence already shows that the product is biosimilar.
Strategic Opportunities: NCE-1 and aBLA Pathways
Beyond the traditional generic and biosimilar pathways, pharmaceutical companies should be aware of emerging strategic opportunities that can provide competitive advantages and expedited market entry.
NCE-1 Opportunities for Chemical Entities
New Chemical Entity (NCE-1) is an application submitted to the U.S. Food and Drug Administration (FDA) for a new drug. This process is also known as a paragraph IV filing. The NCE-1 drug represents a slight modification of an existing approved drug. The term “minus 1” implies that the New Chemical Entity (NCE) is only one small change away from an already FDA-approved and marketed drug.
The approval process for a para IV filing is comparatively quicker and cost-effective than that for an entirely new drug. The time required for the para IV filing is reduced because the FDA may utilize efficacy and safety data available from the original drug.
aBLA Opportunities for Biosimilars
Complex patents: Biologic drugs are protected by multiple patents — covering the molecule, manufacturing process, and formulation. Unlike small-molecule drugs (where Para IV patent challenges are common), biosimilar companies usually handle patents through licensing, settlements, or design modifications.
Interchangeability status: After biosimilar approval, manufacturers can apply for interchangeability, allowing pharmacists to substitute the biosimilar for the original product without the doctor’s approval.
To earn this status, the biosimilar must show that:
- It provides the same clinical result as the reference product for any patient.
- Switching between the biosimilar and the reference product is no riskier than using the reference alone.
Exclusivity benefit: The first interchangeable biosimilar for a reference product gets one year of market exclusivity, giving it a strong competitive edge.
Strategic timing: Unlike Para IV filings (which can trigger a 30-month delay), biosimilar developers must navigate the “patent dance” under the Biologics Price Competition and Innovation Act (BPCIA), a different process that affects when and how they can enter the market.
Why Awareness of These Pathways Matters
Understanding the nuances between generic and biosimilar pathways and recognizing opportunities like NCE-1 and aBLA designations is critical for several reasons:
Strategic Planning
Choosing the right development pathway early influences resource allocation, timeline projections, and ultimate commercial success. A compound that could qualify for NCE-1 status but is developed as a generic represents a missed opportunity for extended market exclusivity.
Competitive Positioning
In the biosimilar space, being first to achieve interchangeability status can provide significant competitive advantages. Similarly, for generics, first-filer Para IV exclusivity can dominate market share during the critical initial generic entry period.
Regulatory Efficiency
Engaging with the FDA early through pre-submission meetings helps identify the most appropriate pathway and avoid costly development missteps. For biologics, this includes Type A, B, C, and Biosimilar Product Development (BPD) meetings that provide critical feedback.
Risk Mitigation
Understanding pathway requirements helps sponsors avoid Refuse-to-File (RTF) actions, which delay market entry and increase development costs. For BLAs, especially, comprehensive gap analyses and regulatory mitigation plans are essential given the extensive documentation requirements.
Navigating Complexity with Expert Guidance
The pharmaceutical regulatory landscape continues to evolve, with increasing complexity in both generic and biosimilar development. Recent regulatory changes, including the full implementation of the Biologics Price Competition and Innovation Act (BPCIA) and ongoing guidance updates from the FDA, require sponsors to stay current with regulatory expectations.
Success in bringing generics and biosimilars to market depends on:
- Early strategic planning to select the optimal regulatory pathway
- Reliable comparator sourcing and compliant supply chain maintenance
- Comprehensive understanding of analytical and clinical requirements
- Robust manufacturing capabilities with appropriate quality controls
- Proactive FDA engagement through advisory meetings and communications
- Experienced in regulatory partnerships to navigate submission complexities
Whether pursuing a traditional ANDA filing with Para IV certification, developing a biosimilar through the 351(k) pathway, or exploring NCE-1 or aBLA opportunities, having expert regulatory guidance throughout the development lifecycle significantly increases the probability of successful approval and timely market entry.
Track NCE-1 opportunities, data exclusivity expiries, Para IV opportunities, and insights into innovator companies in one simple dashboard with the NCE grid.
If you are looking for a reliable comparator drug sourcing solution, contact Spring Bio Solution now.
FAQs
How are biosimilars different from generics?
Biosimilars are large, complex molecules derived from living cells, while generics are small-molecule drugs produced through chemical synthesis. Because biologics naturally show minor batch variations, biosimilars must demonstrate high similarity, not identity. Generics, however, must be chemically identical to their reference drug.
Why is biosimilar approval more complex than generic approval?
Generic approval relies mainly on bioequivalence studies, showing that the generic releases the same active ingredient at the same rate as the reference drug. Biosimilars require extensive analytical, non-clinical, and clinical evidence, including immunogenicity and PK/PD studies, to prove there are no clinically meaningful differences from the reference biologic.
What makes Para IV certification important in generic drug development?
Paragraph IV certification allows a generic manufacturer to challenge patents protecting a brand drug. If successful, the first filer receives 180 days of market exclusivity, creating a significant commercial advantage before other generics can enter the market.
What is interchangeability in biosimilars, and why does it matter?
Interchangeability is a special FDA designation for biosimilars that allows pharmacy-level substitution without prescriber approval. The first interchangeable biosimilar earns one year of exclusivity, offering major competitive benefits. To achieve this status, switching studies must prove no additional risks compared to the reference product.



